
Anwendung des Risikomanagements auf Medizinprodukte
Die dritte Ausgabe der ISO 14971 wurde im Dezember 2019 veröffentlicht. Der Artikel fasst die Norm zusammen und bespricht die Änderungen in der Norm.

Warum eine neue Ausgabe?
Gemäß der ISO ging es bei der neuen Ausgabe der ISO 14971 nicht um eine Überarbeitung des Risikomanagementprozesses, sondern um eine Klarstellung der Umsetzung des Risikomanagementprozesses.[1]
Unternehmen mit Risikomanagementprozessen, die derzeit der Norm entsprechen, sollten also keine großen Überarbeitungen ihrer Prozesse vornehmen müssen, um den geänderten Anforderungen zu entsprechen.
Die Änderungen in der Norm ISO 14971 im Einzelnen
Kapitel 2: Normative References
Die Kapitelstruktur der Norm wurde überarbeitet und an die standardisierten Formatierungsanforderungen der ISO[2] angepasst. Dies führte zur Einführung des neuen Kapitels 2 „Normative Verweise“. Dadurch verschieben sich die weiteren Kapitel um eine Nummer.
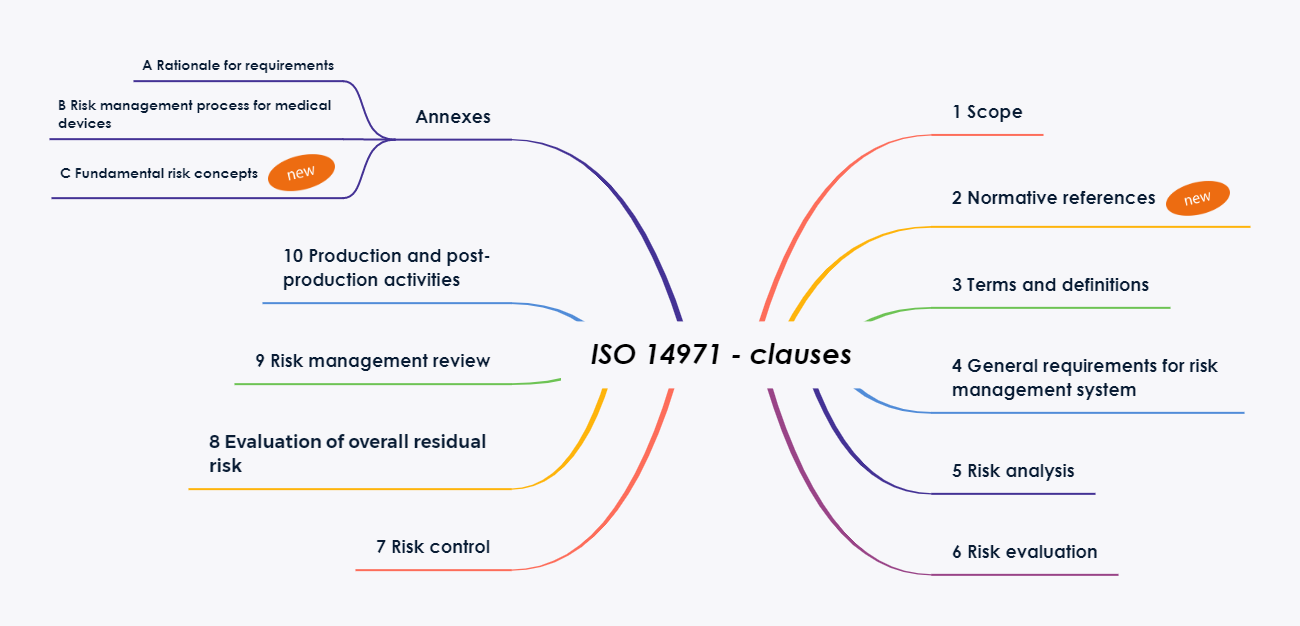
Weiterhin wurde die Anzahl der Anhänge wesentlich verringert. Die meisten der bekannten, erläuternden Anhänge werden in die neue Ausgabe des Technical Report ISO/TR 24971 aufgenommen. Der Report wird voraussichtlich noch in diesem Jahr (geplant 7/2020) veröffentlicht.

Kapitel 3: Terms and Definitions
Im Kapitel 3 werden 4 neue Begriffe definiert:
- Benefit
- Reasonable forseeable misuse
- State of the art
- Harm
Einige weitere Begriffsdefinitionen wurden so angepasst, dass sie inhaltlich den Definitionen aus ISO Guide 63[3] sowie ISO 9000:2015 entsprechen.
Bereits an den neuen Begriffsdefinitionen wird erkennbar, dass der Nutzen des Medizinprodukts mehr Beachtung findet.
„More attention is given to the benefits that are expected from the use of the medical device. The term benefit-risk analysis has been aligned with terminology used in some regulations.“[4]
Kapitel 4: General requirements for risk management system
Hier wird jetzt von Anforderungen an ein Risikomanagement-System gesprochen. Vorher bezogen sich die Anforderungen auf das Risikomanagement.
Weiterhin fällt auf, dass der Risikomanagementplan jetzt auch auf der Abbildung in Kapitel 4.1 explizit genannt wird. Außerdem werden in der Grafik im Kasten „Production and post-production activities“ die neuen Unterkapitel angegeben, die es in der alten Version nicht gab.

4.4 Risk management plan
Bei der Angabe, was der Plan enthalten soll, wird eine Methode für die Evaluation des Gesamtrestrisikos gefordert. Weiterhin sollen Kriterien für die Akzeptanz des Gesamtrestrisikos angegeben werden.
Die Kriterien zur Bewertung der Akzeptanz eines Einzelrisikos können sich also von den Kriterien zur Bewertung der Akzeptanz des Gesamtrestrisikos unterscheiden.
Kapitel 5: Risk analysis
5.1 Risk analysis process
Hier sind nur kleine Änderungen erkennbar. Diese bestehen vorwiegend darin, dass sich Verweise nicht mehr auf die Anhänge, sondern jetzt auf die neue ISO/TR 24971 beziehen.
5.2 Intended use and reasonable forseeable misuse
Dieser Abschnitt wurde überarbeitet und ist jetzt viel klarer formuliert. Er fordert eine dokumentierte Zweckbestimmung (intended use) und führt stichpunktartig den erwarteten Inhalt auf. Weiterhin wird eine Dokumentation des vernünftigerweise vorhersehbaren Missbrauchs gefordert. Dabei wird für die Zweckbestimmung auf die Use-Specification der IEC 62366-1, für den Missbrauch auf die ISO/TR24971 verwiesen.
5.3 Identification of characteristics related to safety
Die Forderung nach der Identifizierung sicherheitsbezogener Merkmale war in der alten Ausgabe noch im Kapitel 5.2 genannt worden. Sie wurde jetzt in einen eigenen Abschnitt 5.3 ausgelagert. Die bekannte Frageliste aus dem Anhang C ist jetzt Teil der ISO/TR 24971.
Interessant ist die Anmerkung 2, die auch den Begriff „essential performance“ aus der IEC 60601-1 einbezieht.
5.4 Identification of hazards and hazardous situations
Der Abschnitt 5.4 fasst zum einen die Unterkapitel 4.3 und 4.4 der alten Ausgabe zusammen. Zum anderen wird der Aspekt der Risikoabschätzung in ein eigenes Unterkapitel (5.5) ausgelagert.
Dabei sollen ausgehend von der Zweckbestimmung, dem vernünftigerweise vorhersehbaren Missbrauch und den sicherheitsbezogenen Merkmalen Gefährdungen identifiziert werden. Es müssen sowohl Normal- als auch Fehlerbedingungen berücksichtigt werden.
Anschließend fordert die Norm, dass für jede identifizierte Gefährdung vernünftigerweise vorhersehbare Abfolgen oder Kombinationen von Ereignissen berücksichtigt werden und die sich so ergebenden Gefährdungssituationen aufgezeichnet werden müssen.
5.5 Risk estimation
Für jede identifizierte Gefährdungssituation soll das Risiko abgeschätzt werden. Dabei wurden die Anforderungen der alten Ausgabe übernommen.
Kapitel 6: Risk evaluation
Hier gibt es nichts Neues. Weiterhin muss die Risikobeherrschung (Kapitel 7.1 bis 7.5) nicht für akzeptable Risiken angewendet werden. Wie die EU das sieht, ist mir hier noch nicht klar – denken Sie auch gerade an die Anhänge Z der EN ISO 14971:2012?
Kapitel 7: Risk control
Auch hier hat sich die Kapitelstruktur geändert. Im Wesentlichen fällt nur der inhaltlich wenig ergiebige Abschnitt 6.1 weg.
7.1 Risk control option analysis
Hier fällt auf, dass die Optionen zur Risikobeherrschung überarbeitet wurden:
Ergänzt wurde in der ersten Option die Einbeziehung der Herstellung:
„inherently safe design and manufacture„
In der dritten Option wurde das Anwendertraining ergänzt:
„information for safety and, where appropriate, training to users“
Weiterhin wurde ergänzt:
„Relevant standards should be applied as part of the risk control option analysis.“
Dies wurde bisher nur in einer Anmerkung verdeutlicht.
7.2 Implementation of risk control measures
In diesem Abschnitt wurden vor allem Anmerkungen und zwei Beispiele ergänzt, die sich auf die Verifikation der Wirksamkeit der Maßnahmen beziehen. Dabei wird explizit auf Process- und Design-Qualification eingegangen.
7.3 Residual risk evaluation
Hier fällt der redundante Abschnitt über die Offenlegung akzeptabler Restrisiken weg.
7.4 Benefit-risk analysis
Hier gibt es keine wesentlichen Änderungen.
7.5 Risks arising from risk control measures
Hier gibt es keine wesentlichen Änderungen.
7.6 Completeness of risk control
Hier gibt es keine wesentlichen Änderungen.
Kapitel 8: Evaluation of overall residual risk
Im Gegensatz zur alten Ausgabe soll für die Bewertung des Gesamtrestrisikos neben allen Einzelrisiken auch eine Bewertung des Restrisikos gegenüber dem Nutzen des Medizinprodukts gemäß seiner Zweckbestimmung erfolgen.
Dabei sollen für die Bewertung des Gesamtrestrisikos die Akzeptanzkriterien aus dem Risikomanagementplan herangezogen werden. Neu ist, dass es unterschiedliche Akzeptanzkriterien für die Einzelrisiken und für das Gesamtrestrisiko geben kann.
Wenn das Gesamtrestrisiko unakzeptabel ist, kann der Hersteller erwägen,
- weitere Maßnahmen zur Risikokontrolle einzuführen, um das Restrisiko weiter zu verringern oder
- das Medizinprodukt oder die Zweckbestimmung des Medizinprodukts zu modifizieren.
Ist das nicht möglich, bleibt das Gesamtrestrisiko unakzeptabel.
Wenn das Gesamtrestrisiko als akzeptabel beurteilt wird, muss der Hersteller die Benutzer über signifikante Restrisiken informieren und die erforderlichen Informationen in die Begleitdokumentation aufnehmen, um diese Restrisiken offenzulegen.
Kapitel 9: Risk management review
Der Titel des Kapitels hat sich zwar geändert: In der alten Ausgabe stand hier noch „Risk management report“. Inhaltlich bleibt aber alles beim Alten.
Kapitel 10: Production and post-production activities
Dieses Kapitel wurde umfassend überarbeitet. Das erkennt man schon am Titel des Kapitels: Nicht die Informationen für die nachgelagerten Phasen stehen im Fokus, sondern die aus Informationssammlung und -bewertung durchzuführenden Maßnahmen.
Weiterhin wurde dieser Abschnitt hinsichtlich des Kapitel 8 der ISO 13485 angepasst, das sich mit der Bearbeitung von Beschwerden, Berücksichtigung von Kundenfeedback, internen Audits, Kontrolle fehlerhafter Produkte, Datenanalyse und Verbesserungen befasst.
10.1 General
Dieser Abschnitt betont, dass der Hersteller ein System etablieren muss, das aktiv relevante Informationen aus Produktion und den Postproduktionsphasen sammelt und bewertet, um die richtigen Aktivitäten daraus abzuleiten.
10.2 Information collection
Hier wird ausführlich beschrieben, welche Informationsquellen zu berücksichtigen sind.
10.3 Information review
Hier wird erläutert, wie die Informationen bezüglich ihrer Sicherheitsrelevanz zu überprüfen sind.
10.4 Actions
Der neue Abschnitt „Actions“ beschreibt, welche Maßnahmen anzuwenden sind, wenn festgestellt wird, dass Informationen sicherheitsrelevante Aspekte des Medizinprodukts betreffen.
Dabei wird zum einen auf Aspekte des jeweiligen Medizinprodukts eingegangen, z. B.:
- Überprüfung der Risikomanagementakte
- Überprüfung der Akzeptanz des Restrisikos
- Maßnahmen, die sich auf im Markt befindliche Produkte beziehen
Zum anderen wird auf den gesamten Risikomanagementprozess eingegangen. Beispielsweise
- auf Überprüfung der Eignung des RM-Prozesses und
- darauf, welche Änderungen am Prozess durchgeführt werden müssen.
Annex A Rational for Requirements
Dieser Anhang erläutert die im normativen Teil beschriebenen Anforderungen der ISO 14971.
Annex B Risk management process for medical devices
Anhang B enthält eine Tabelle der Entsprechungen zwischen den Normen von 2007 bis 2019, sowie ein Übersichtsdiagramm des
Risikomanagementprozesses für Medizinprodukte.
Annex C Fundamental risk concepts
Anhang C beschäftigt sich mit den Begriffen Hazard, Sequence of events, Hazardous situations, Harm und Risk und bringt Beispiele für die genannten Begriffe. In der vorherigen Ausgabe fand man dieses Material im Anhang E. Der ursprüngliche Anhang C wird in die ISO TR 24971 verschoben.
Fazit:
Die neue ISO 14971 ist klarer formuliert und gibt bessere Vorgaben zu den einzelnen Anforderungen. Auch die Struktur der Norm und die Einteilung der Unterkapitel wurde verbessert. Allerdings muss man die vorher sehr schlanke TR 24971 (12 Seiten) nun zusätzlich erwerben, um den vollständigen Umfang der alten ISO 14971 zu erhalten.
Inhaltlich sind nur wenige Änderungen durchgeführt worden. Dazu zählen insbesondere:
- Besondere Betonung der Risiko-Nutzen-Abwägung
- Ergänzung des Planes um Methode und Kriterien für die Gesamtrisikobewertung (Kapitel 4.4)
- Durchführung der Bewertung des Gesamtrestrisikos (Kapitel 8)
- Anforderungen an die aktive Sammlung und Bewertung der Informationen aus den nachgelagerten Phasen sowie Betonung der durchzuführenden Maßnahmen (Kapitel 10)
Das Ziel der ISO, dass etablierte Risikomanagementprozesse nur wenig Änderungen bedürfen, um den Anforderungen der neuen Ausgabe der Norm ISO 14971 zu entsprechen, wurde aus meiner Sicht erreicht. Die Klarstellungen helfen den Herstellern dabei, die Anforderungen leichter normgerecht umzusetzen.
- https://www.meddeviceonline.com/doc/a-look-at-the-iso-and-iso-tr-updates-0001 ↩
- verantwortlich für diese Anforderungen ist das ISO Technical Management Board. ↩
- ISO/IEC Guide 63:2019 Guide to the development and inclusion of aspects of safety in International Standards for medical devices ↩
- aus EN ISO 14971:2019, Foreword ↩