
Wer sich mit Medizintechnik beschäftigt, sollte auch die Gesetze und Richtlinien kennen, die dafür notwendig sind. Daher möchte ich heute auf die Rechtslage in Europa eingehen. Wenn Sie beim Gedanken an MDD, MPG und grundlegende Anforderungen mit Unbehagen reagieren, dann sollten Sie den Artikel lesen. Zudem erfahren Sie hier, was Kondome und Defibrillatoren gemeinsam haben.
Was ist ein Medizinprodukt?
Medizinprodukte sind in der Richtlinie 93/42/EWG definiert als:
Alle einzeln oder miteinander verbunden verwendeten Instrumente, Apparate, Vorrichtungen, Software, Stoffe oder anderen Gegenstände, einschließlich der vom Hersteller speziell zur Anwendung für diagnostische und/oder therapeutische Zwecke bestimmten und für ein einwandfreies Funktionieren des Medizinprodukts eingesetzten Software, die vom Hersteller zur Anwendung für Menschen für folgende Zwecke bestimmt sind:
- Erkennung, Verhütung, Überwachung, Behandlung oder Linderung von Krankheiten;
- Erkennung, Überwachung, Behandlung, Linderung oder Kompensierung von Verletzungen oder Behinderungen;
- Untersuchung, Ersatz oder Veränderung des anatomischen Aufbaus oder eines physiologischen Vorgangs;
- Empfängnisregelung
und deren bestimmungsgemäße Hauptwirkung im oder am menschlichen Körper weder durch pharmakologische oder immunologische Mittel noch metabolisch erreicht wird, deren Wirkungsweise aber durch solche Mittel unterstützt werden kann.
Sofern die Zweckbestimmung (engl. Intended Use) Ihres Produktes für diese Zwecke gedacht ist, so handelt es sich um ein Medizinprodukt. Dabei kann auch Software (Medical Apps oder Webseiten) als Medizinprodukt gelten.
MDD und MPG
Für Medizinprodukte gibt es in Europa drei Richtlinien, die die rechtlichen Rahmenbedingungen definieren:
- Richtlinie 93/42/EWG über Medizinprodukte (engl. Medical Device Directive – MDD)
- Richtlinie 98/79/EG über In-vitro-Diagnostika (IVDD)
- Richtlinie 90/385/EWG über aktive implantierbare medizinische Geräte (AIMDD)
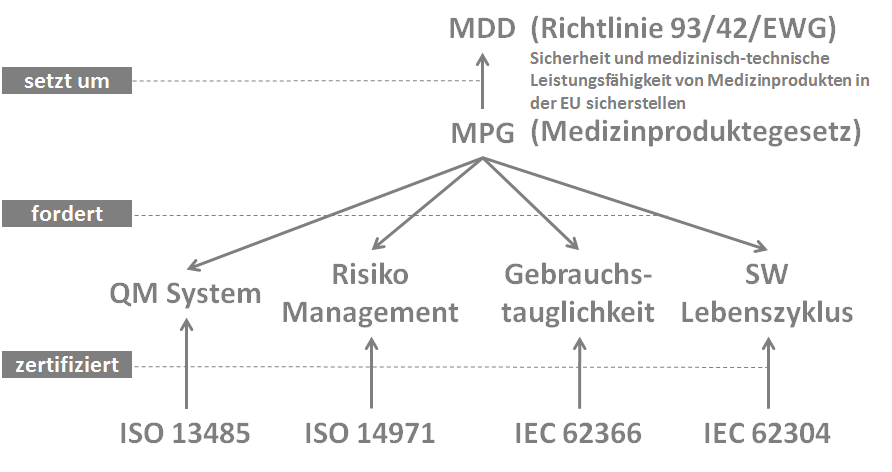
Diese drei Richtlinien sollen die Sicherheit und die medizinisch-technische Leistungsfähigkeit von Medizinprodukten in der EU sicherstellen. Die Mitgliedsstaaten der EU sind dazu verpflichtet alle erforderlichen Maßnahmen zu treffen, damit die Produkte nur in Verkehr gebracht und/oder in Betrieb genommen werden dürfen, wenn sie bei sachgemäßer Lieferung, Installation, Instandhaltung und ihrer Zweckbestimmung entsprechender Verwendung die Anforderungen dieser Richtlinien erfüllen.
Die Umsetzung der Medizinprodukterichtlinie in jeweils nationales Recht erfolgt durch nationale Gesetze. In Deutschland durch das Medizinproduktegesetz (MPG).
|
|
| Dipl.-Ing. Goran Madzar, Gesellschafter, Senior Systems Engineer E-Mail: madzar@medtech-ingenieur.de Tel.: +49 9131 691 240 |
|
Benötigen Sie Unterstützung bei der Entwicklung Ihres Medizingeräts? Wir helfen gerne! Die MEDtech Ingenieur GmbH bietet Hardware-Entwicklung, Software-Entwicklung, Systems Engineering, Mechanik-Entwicklung und Beratung aus einer Hand. Nehmen Sie Kontakt mit uns auf. |
|
Für Medizinproduktehersteller ergeben sich aus dem MDD vier wesentliche Anforderungen zu den Bereichen:
- Qualitätsmanagement
- Risikomanagement
- Software-Lebenszyklus-Prozess
- Gebrauchstauglichkeit
Zu diesen vier Themenbereichen gibt es harmonisierte europäische Normen:
- EN ISO 13485 Medizinprodukte – Qualitätsmanagementsysteme – Anforderungen für regulatorische Zwecke
- EN ISO 14971 Anwendung des Risikomanagements auf Medizinprodukte
- EN 62304 Medizingeräte-Software – Software-Lebenszyklus-Prozesse
- EN 62366 Anwendung der Gebrauchstauglichkeit auf Medizinprodukte
Harmonisiert bedeutet, dass bei Einhaltung dieser harmonisierten Norm ebenfalls die wesentlichen Anforderungen der anwendbaren Richtlinie erfüllt sind. Diese Annahme wird als „Konformitätsvermutung“ bezeichnet. Harmonisierte Normen entstehen in Zusammenarbeit der Europäischen Kommission mit europäischen Normungsgremien (CEN oder CENELEC).
Klassifizierung von Medizinprodukten
Man unterscheidet aktive und nicht aktive Medizinprodukte. Aktive Medizinprodukte nutzen eine externe Energiequelle (Strom, Akku, Batterie, thermische oder kinetische Energie oder Gasdruck). Nicht aktive Medizinprodukte sind „passiv“ oder werden mit Muskelkraft oder Schwerkraft betrieben.
Abhängig vom Risiko der Anwendung werden die Medizinprodukte in vier Klassen eingeteilt: I, IIa, IIb und III.
Die Klassen sind EU-weit durch den Anhang IX der Richtlinie 93/42/EWG festgelegt:
- Klasse I
- Keine methodischen Risiken
- geringer Invasivitätsgrad
- kein oder unkritischer Hautkontakt
- vorübergehende Anwendung ≤ 60 Minuten
- Klasse IIa
- Anwendungsrisiko
- mäßiger Invasivitätsgrad
- kurzzeitige Anwendungen im Körper (im Auge, intestinal, in chirurgisch geschaffenen Körperöffnungen)
- kurzzeitig ≤ 30 Tage, ununterbrochen oder wiederholter Einsatz des gleichen Produktes
- Klasse IIb
- Erhöhtes methodisches Risiko
- systemische Wirkungen
- Langzeitanwendungen
- nicht invasive Empfängnisverhütung
- langzeitig ≥ 30 Tage, sonst wie bei kurzzeitig
- Klasse III entspricht hohem Gefahrenpotential
- Besonders hohes methodisches Risiko
- zur langfristigen Medikamentenabgabe
- Inhaltsstoff tierischen Ursprungs und im Körper
- unmittelbare Anwendung an Herz, zentralem Kreislaufsystem oder zentralem Nervensystem
- und natürlich invasive Empfängnisverhütung
Beispiele von Medizinprodukten zu den verschiedenen Klassen sind in der Tabelle unten dargestellt.
| Klasse I | Klasse IIa | Klasse IIb | Klasse III |
| ärztliche Instrumente | Dentalmaterialien | Anästhesiegeräte | Herzkatheter |
| Gehilfen | Desinfektionsmittel (für Instrumente und Geräte) | Beatmungsgeräte | künstliche Hüft-, Knie-, oder Schultergelenke |
| Rollstühle | diagnostische Ultraschallgeräte | Bestrahlungsgeräte | Stents |
| Pflegebetten | Einmalspritzen | Blutbeutel | resorbierbares chirurgisches Nahtmaterial |
| Stützstrümpfe | Hörgeräte | Defibrillatoren | Intrauterinpessar (Spirale) |
| Verbandmittel | Kontaktlinsen | Dialysegeräte | Brustimplantat |
| wiederverwendbare chirurgische Instrumente | PACS | Kondome | Herzschrittmacher |
| OP-Textilien | Trachealtuben | Kontaktlinsenreiniger |
Die Klassifizierung erfolgt auf Grundlage des bestimmungsgemäßen Gebrauchs (engl. intended use). Wussten Sie, dass Kondome und Defibrillatoren gleich klassifiziert sind? Ich finde das schon ein wenig amüsant.
Konformitätsbewertung
Die Konformitätsbewertung wird durch den Hersteller vorgenommen. Dabei spielt die Risikoklasse für die Konformitätsbewertung eine wichtige Rolle. Für ein Klasse I Produkt ist eine Konformitätserklärung nach Anhang VII ausreichend. Dazu ist der Hersteller verpflichtet, mittels einer technischen Dokumentation den Nachweis der grundlegenden Anforderungen zu erbringen. Der Hersteller muss auch in der Lage sein, nach dem Inverkehrbringen das Produkt zu beobachten und Korrekturen zu veranlassen. Dabei ist die Einbeziehung der Benannten Stelle nicht nötig. Anders sieht es bei den höheren Risikoklassen (IIa, IIb und III) aus. Hier kann ein Konformitätsbewertungsverfahren nach Anhang II erfolgen. Dabei muss ein Hersteller ein vollständiges Qualitätssicherungssystem (meistens nach ISO 13485) aufbauen. Dieses wird durch Benannte Stellen in Audits regelmäßig zertifiziert und überprüft. Eine andere Möglichkeit besteht darin die Entwicklung über eine EG-Baumusterprüfung nach Anhang III durchführen zu lassen. Die Benannte Stelle prüft mittels eines Baumusters sowie den dazugehörigen Dokumenten die Erfüllung der grundlegenden Anforderungen. Anschließend muss sichergestellt werden, dass der Hersteller in der Lage ist, das Produkt korrekt herzustellen. Dies kann durch eine EG-Prüfung nach Anhang IV erfolgen, bei der die fertigen Produkte der Benannten Stelle zur Prüfung vorgelegt werden. Es ist auch möglich die Qualitätssicherung der Produktion nach Anhang V oder die Qualitätssicherung des Produktes nach Anhang VI durch eine Benannte Stelle zertifizieren zu lassen.
CE-Kennzeichnung
Nach einem erfolgreichen Konformitätsbewertungsverfahren darf eine CE-Kennzeichnung auf das Produkt aufgebracht und somit in Europa in Verkehr gebracht werden. Ist eine Benannte Stelle beim Konformitätsbewertungsverfahren beteiligt, so wird deren vierstellige Kennnummer hinter der Kennzeichnung angegeben.
Benannte Stellen (engl. Notified Bodies) sind staatlich autorisierte Stellen, die – abhängig von der Risikoklasse der Medizinprodukte – Prüfungen und Bewertungen im Rahmen der vom Hersteller durchzuführenden Konformitätsbewertung durchführen und deren Korrektheit nach einheitlichen Bewertungsmaßstäben bescheinigen. Hersteller können sich an eine Benannte Stelle ihrer Wahl wenden, die für das entsprechende Verfahren und die betreffende Produktkategorie benannt ist.
Fazit
Ich hoffe, dass Ihnen die Gesetze und Richtlinien ein bisschen klarer geworden sind. Der Artikel hat nicht den Anspruch alle Themen vollständig und umfänglich zu behandeln. Ziel war es, die grundlegenden Zusammenhänge zu erläutern. Für diesen Artikel gilt daher: „Zu Risiken und Nebenwirkungen lesen Sie die Richtlinien und Gesetze oder wenden Sie sich an Ihren Qualitätsmanager :-) “
Ich freue mich über Feedback und wenn Sie mit mir in Kontakt treten. Sie können gerne auch einen Kommentar zu dem Artikel abgeben. Falls Sie jemanden kennen, für den der Blog ebenfalls interessant sein könnte, freue ich mich auch sehr über eine Weiterempfehlung.
Viele Grüße
Goran Madzar