
Referenzen:
Wenn im Folgenden von IEC 62304 oder ISO 14971 gesprochen wird, sind folgende Ausgaben gemeint: IEC 62304:2006 + A1:2015 und EN ISO 14971:2012
Problemstellung:
Wir entwickeln eine Software/Firmware, welche Bestandteil eines Medizinprodukts ist. Die Software (bzw. das Softwaresystem) wird der Software-Sicherheitsklasse B oder C zugeordnet. Diese Zuordnung hat u.a. zur Folge das gemäß IEC 62304 ein Software Risikomanagement Prozess (Kapitel 7) gefordert ist.
Anspruch dieses Artikels:
Der folgende Artikel soll einen praktischen Ansatz darstellen, wie ein Software Risikomanagement Prozess dokumentiert werden kann und welche Methoden geeignet sind, mögliche Software Fehler zu identifizieren.
Methoden zur Identifizierung
Wie (leider) in vielen Fällen gibt die IEC 62304 keine konkreten Vorgaben welche Methodik zur Identifikation möglicher Software Fehler verwendet werden soll. In einigen Unternehmen werden Methoden wie etwa die FMEA (dt. Fehlzustandsart- und -auswirkungsanalyse) angewendet, da diese in der ISO14971 zusammen mit dem Risikomanagement erwähnt werden. Doch inwiefern eignen sich diese Methoden für Software-Risikomanagement?
Exkurs: FMEA und FMECA
Die FMEA ist als Analysetechnik für die Funktionsfähigkeit von Systemen in der IEC 60812 beschrieben. Die Technik wird branchenübergreifend angewendet und ist nicht spezifisch für die Medizinprodukte-Industrie. Genauer hat die FMEA ihren Ursprung im amerikanischen Militär und ist heute v.a. in der Automobilindustrie weit verbreitet.
Wird die FMEA um die Ausfallbedeutungsanalyse ergänzt, dann spricht man von einer FMECA (engl. failure modes, effects and criticality analysis)
DIE EINE und WAHRE FMEA gibt es vermutlich nicht. Jeder Industriezweig oder Firma nutzt die FMEA auf die für sich geeignete Weise. Jedoch vielen ist gemein, eine Risikoprioritätszahl (RPZ) zu verwenden.
Die Risikoprioritätszahl setzt sich in der deutschsprachigen Literatur aus den drei folgenden Komponenten zusammen:
- B: Schwere/Bedeutung
- E: Entdeckungswahrscheinlichkeit
- A: Auftretenswahrscheinlichkeit
Die IEC 60812 spricht weiter auch von einer Risikoakzeptanzbeurteilung. Das kommt uns bekannt vor und darauf werden wir später noch einmal genauer eingehen.
Im Anhang der IEC 60812 gibt es Beispiele für die Erstellung einer FMEA bzw. FMECA. Auch dieses Schema kommt uns aus der Welt der ISO 14971 bekannt vor.

Forderungen der IEC 62304 bzgl. Software Risiko Management
Kehren wir nun zurück zu den Medizinprodukten. Die IEC 62304 schreibt einleitend:
„Software ist oft ein integraler Bestandteil der MEDIZINPRODUKTE-Technik. Um die SICHERHEIT und Wirksamkeit eines MEDIZINPRODUKTS, das Software enthält, herzustellen, bedarf es der Kenntnis darüber, was die Software bewirken soll, und des Nachweises darüber, dass die Software diese Wirkung erzielt, ohne unvertretbare RISIKEN zu verursachen.“
Den auch in anderen Bereichen (Qualitätsmanagementsystem, Klassifizierung der Medizinprodukte) bekannten risikobasierten Ansatz folgt die IEC 62304 weiter. Die Methodik dahinter ist die Software Sicherheitsklassifizierung, welche die Produkte in drei Klassen teilt: A, B, C.

Basierend auf der Einteilung der Software-Sicherheitsklassifizierung erfolgt die Tiefe der Dokumentation und Verifikationstätigkeiten. Hinweis: Dies soll einen Überblick darstellen. Der Artikel geht an dieser Stelle nicht tiefer auf das Thema ein, dies würde ein ganz eigene Diskussion nach sich ziehen.
Für Sicherheitsklasse B und C wird gefordert, dass ein Software Risikomanagement geplant wird. Für beide Klassen wird weiter gefordert, dass die Identifizierung und Vermeidung gemeinsamer Software Fehler geplant wird, die sogenannten common software defects.
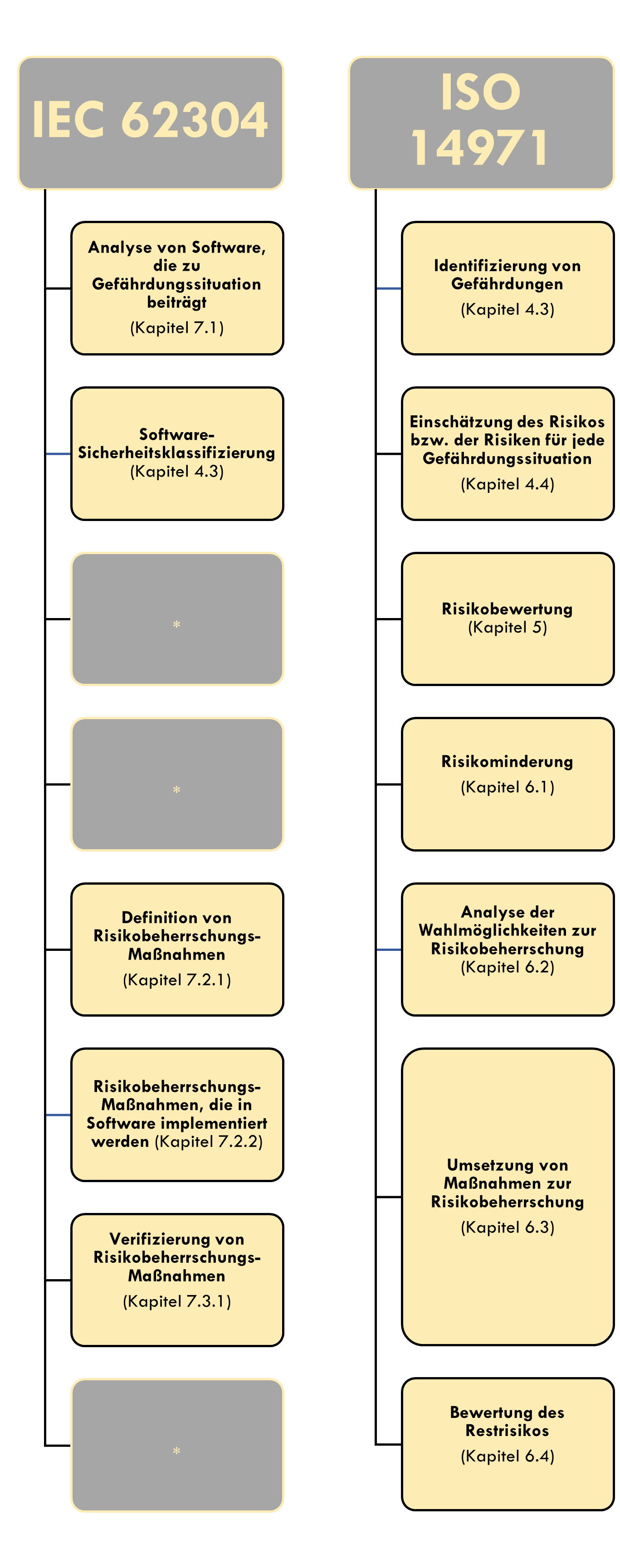
Abgrenzung der IEC 62304 und der ISO 14971
Weiter wichtig: Umgang mit Software Änderungen. Wird hier bewusst nicht betrachtet.
Es ist zu erkennen, dass die in der ISO 14971 geforderten Aspekte der Risikobewertung in der IEC 62304 nicht vorkommen / gefordert sind.
Um es an dieser Stelle bereits vorweg zu nehmen: Dies ist auch der entscheidende Grund warum wir den Einsatz einer FMEA bzw. FMECA für nicht sinnvoll erachten. Denn der Einsatz einer RPZ in der FMEA ist schwierig zu begründen. Fällt die RPZ heraus, bleibt unserer Einschätzung nicht mehr viel übrig was den Einsatz einer FMEA begründet. Daher sehen wir es als sinnvoll an, die Software Risikoanalyse mit in die Systemrisikoanalyse gemäß 14971 zu integrieren. Wie wir das praktisch umsetzen, zeigen wir gleich.
Die Schwierigkeit mit der RPZ:
Wie oben erwähnt ergibt sich die RPZ aus der Schwere und P1 und P2 (in manchen Fällen auch nur P, wenn die Wahrscheinlichkeit kombiniert betrachtet wird).
In einigen FMEAs kommt man dann auf Werte für RPZ im Bereich RPZ = 10 * 10 * 10 = 1000. Häufig angewendet wird eine Risikoakzeptanzbeurteilung von < 100 und >= 100. Doch wie ist dies zu begründen? In den meisten Fällen fehlt die Evidenz und die Einstufung wirkt willkürlich, oder die Einstufung wird benutzt, um zu entscheiden, bis zu welcher Zahl die Maßnahmen durchgeführt werden und passt nicht zum As Low As Possible Prinzip.
Mit der ISO 14971 dagegen hat man eine sehr gute Methode an der Hand mit der wir, wie gewohnt, eine Risikomatrix aufstellen können. Die darin aufgeteilte Einschätzung des Risikos anhand der Darstellung von Schwere und Auftretenswahrscheinlichkeit begründet sich auf den im Risikomanagement und der klinischen Bewertung verfügbaren Daten.
Diese Daten können sein:
- Normen
- wissenschaftlich-technischen Daten;
- Marktdaten von ähnlichen, bereits in Anwendung befindlichen Medizinprodukten einschließlich veröffentlichter Berichte über Vorkommnisse
- Gebrauchstauglichkeitsuntersuchungen mit typischen Anwendern
- klinische Daten
- Ergebnissen geeigneter Untersuchungen
- Gutachten
- externen Qualitätsbeurteilungsprogrammen
Unsere Einschätzung sehen wir auch durch die Erläuterung im Anhang der IEC 62304 bestärkt:
„Das RISIKOMANAGEMENT von Software ist ein Teil des RISIKOMANAGEMENTS des gesamten MEDIZINPRODUKTS und kann vernünftigerweise nicht isoliert betrachtet werden.“
Aus den vorangegangenen Punkten kommen wir zu dem Schluss, dass alle Einträge einer „FMEA“, sprich mögliche Fehler und dazugehörige Ursachen, unabhängig einer RPZ als wertvoll anzusehen sind. Daher dient die „FMEA“ nur der reinen Identifikation solcher Elemente und behandelt alle gleichwertig. Es erfolgt keine quantitative Bewertung.
Anmerkung: Dies ist eine subjektive Einschätzung aus den Entwicklungsprojekten und erhebt nicht den Anspruch generell den Einsatz einer FMEA in Frage zu stellen. In einigen anderen Bereichen wie beispielsweise Produktion, gibt es eine ganz andere Sichtweise auf die Dinge. Wer in seinem Projekt und Audits gute Erfahrungen damit gemacht hat, sollte an seiner Strategie festhalten. Wir freuen uns aber, wenn der Artikel an dieser Stelle zum Nachdenken anregt und die ein oder andere Diskussion anstößt.
Beispiel einer möglichen Dokumentation eines Software Risiko Management Prozesses
Die Art und Weise wie dokumentiert wird, hängt im Wesentlichen vom zugrundeliegenden Qualitätsmanagementsystem, Entwicklungs- und Softwareentwicklungsprozess sowie dem Risikomanagement-Prozess ab.
1. Überblick über die notwendige Dokumentation in Zusammenhang mit SW Risk Management

2. Risiko Management Akte / Risikoanalyse Tabelle
![]()
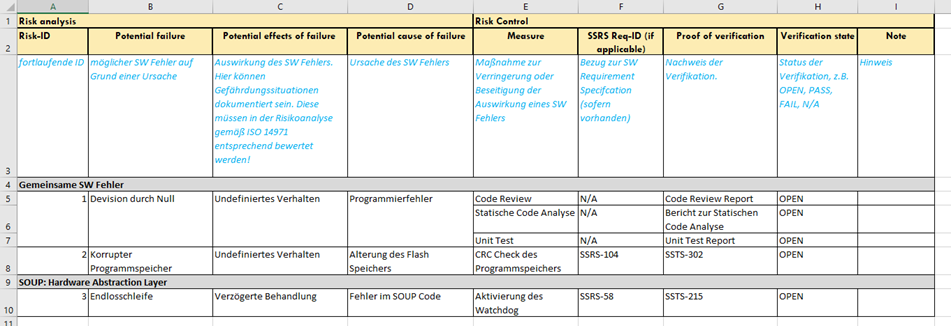
Stellt sich noch die Frage wie oft wir das oben genannte Schema anwenden. Sollte man die Gefährdungen für jede SW Einheit betrachten oder nur einmal für das SW System? Die Antwort hierauf ergibt sich durch die SW Architektur. Läuft mein System auf lediglich einer Hardware und ist die Architektur entsprechend übersichtlich, würden wir nicht empfehlen das SW Risikomanagement auf mehrere Komponenten aufzuteilen. Schließlich sind die Maßnahmen, vorausgesetzt die SW Sicherheitsklassen sind gleich, in der Regel identisch.
Erstreckt sich das SW System aber auf mehrere, komplexere SW Systeme, auf unterschiedlicher Hardware, empfiehlt es sich diese Komponenten getrennt zu betrachten. Schließlich können sich bei so einer Architektur, verschiedene Effekte aus den gleichen SW Fehlern ergeben.