
Application of risk management to medical devices
The third edition of ISO 14971 was published in December 2019. This article summarizes the standard and discusses the changes in the standard.

Why a new edition?
According to ISO, the new edition of ISO 14971 was not about revising the risk management process, but rather about clarifying the implementation of the risk management process.[1]
Companies with risk management processes that currently comply with the standard should therefore not have to make major revisions to their processes to comply with the changed requirements.
The changes in the ISO 14971 standard in detail
Chapter 2: Normative References
The chapter structure of the standard has been revised and adapted to the standardized formatting requirements of ISO[2] This led to the introduction of the new Chapter 2 “Normative references”This shifts the remaining chapters by one number.
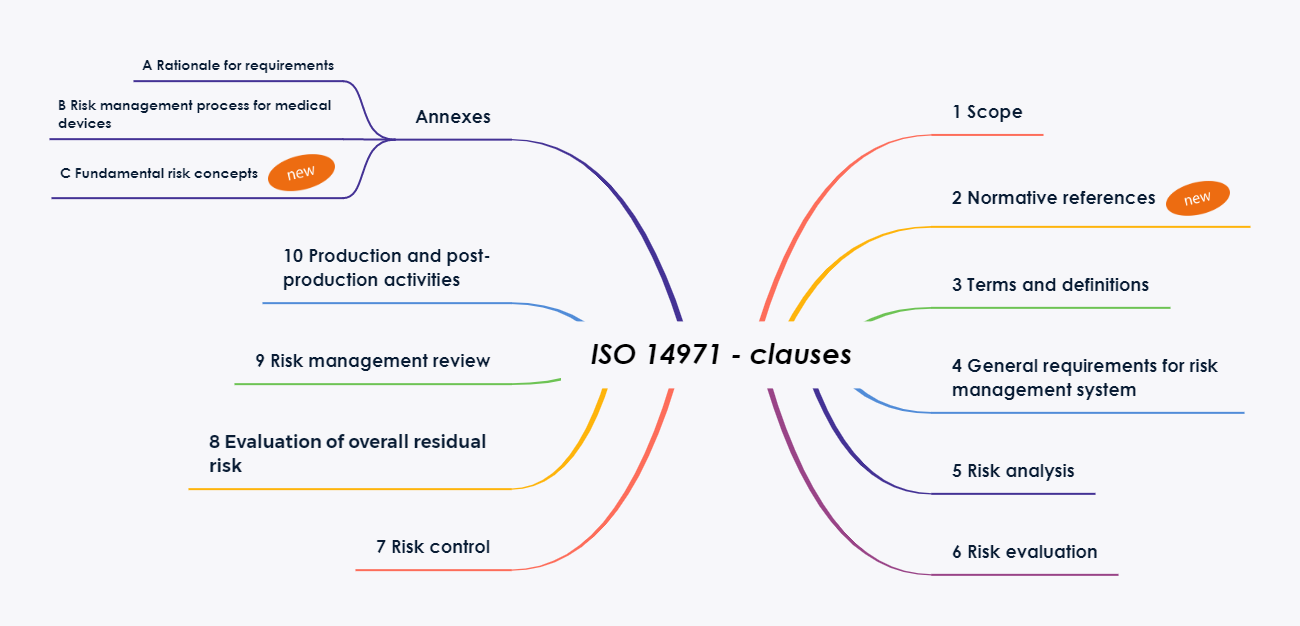
Furthermore, the number of annexes has been significantly reduced. Most of the familiar explanatory annexes will be included in the new edition of Technical Report ISO/TR 24971. The report is expected to be published later this year (planned for July 2020).

Chapter 3: Terms and Definitions
In Chapter 3, four new terms are defined:
- Benefit
- Reasonable forseeable misuse
- State-of-the-art
- Harm
Some other definitions have been adapted so that they correspond in content to the definitions in ISO Guide 63[3] as well as ISO 9000:2015 are equivalent to.
The new definitions already show that the benefits of the medical device are receiving more attention.
"More attention is given to the benefits that are expected from the use of the medical device. The term benefit-risk analysis has been aligned with terminology used in some regulations."[4]
Chapter 4: General requirements for risk management system
Here we are now talking about requirements for a risk managementsystem Previously, the requirements related to risk management.
It is also noticeable that the risk management plan is now explicitly mentioned in the figure in Chapter 4.1. Furthermore, the graphic in the box “Production and post-production activities” the new subchapters that were not present in the old version.

4.4 Risk management plan
When specifying what the plan should contain, a method for evaluating the overall residual risk is required. Furthermore, criteria for the acceptance of the overall residual risk should be specified.
The criteria for assessing the acceptance of an individual risk may therefore differ from the criteria for assessing the acceptance of the overall residual risk.
Chapter 5: Risk analysis
5.1 Risk analysis process
Only minor changes are noticeable here. These mainly consist of references no longer referring to the annexes, but now to the new ISO/TR 24971.
5.2 Intended use and reasonable forseeable misuse
This section has been revised and is now much clearer. It requires a documented intended use and lists the expected content in bullet points. Furthermore, documentation of reasonably foreseeable misuse is required. For the intended use, reference is made to the use specification of IEC 62366-1, and for misuse, to ISO/TR24971.
5.3 Identification of characteristics related to safety
The requirement to identify security-related features was mentioned in Chapter 5.2 of the previous edition. It has now been moved to a separate Section 5.3. The familiar list of questions from Annex C is now part of ISO/TR 24971.
Note 2 is interesting, which also uses the term “essential performance” from IEC 60601-1.
5.4 Identification of hazards and hazardous situations
Section 5.4 summarizes subchapters 4.3 and 4.4 of the previous edition. Furthermore, the risk assessment aspect is moved to a separate subchapter (5.5).
Hazards should be identified based on the intended purpose, reasonably foreseeable misuse, and safety-related characteristics. Both normal and fault conditions must be considered.
The standard then requires that for each identified hazard, reasonably foreseeable sequences or combinations of events must be taken into account and the resulting hazardous situations must be recorded.
5.5 Risk estimation
For each identified hazard situation, the risk should be assessed. The requirements of the previous edition have been adopted.
Chapter 6: Risk evaluation
There's nothing new here. Furthermore, risk control (chapters 7.1 to 7.5) does not need to be applied for acceptable risks. I'm not yet clear on how the EU views this – are you also thinking about Annex Z of EN ISO 14971:2012?
Chapter 7: Risk control
Here, too, the chapter structure has changed. Essentially, only section 6.1, which is less informative, has been omitted.
7.1 Risk control option analysis
It is noticeable here that the risk management options have been revised:
The first option added the inclusion of manufacturing:
"inherently safe design and manufacture„
In the third option, user training was added:
“information for safety and, where appropriate, training to users”
Furthermore, it was added:
“Relevant standards should be applied as part of the risk control option analysis.”
This has so far only been clarified in one note.
7.2 Implementation of risk control measures
This section primarily includes comments and two examples related to verifying the effectiveness of the measures. Process and design qualification are explicitly addressed.
7.3 Residual risk evaluation
The redundant section on the disclosure of acceptable residual risks is omitted here.
7.4 Benefit-risk analysis
There are no significant changes here.
7.5 Risks arising from risk control measures
There are no significant changes here.
7.6 Completeness of risk control
There are no significant changes here.
Chapter 8: Evaluation of overall residual risk
In contrast to the old edition, the assessment of the overall residual risk should include, in addition to all individual risks, an assessment of the residual risk compared to the benefit of the medical device in accordance with its intended purpose.
The acceptance criteria from the risk management plan should be used to assess the overall residual risk. What's new is that there can be different acceptance criteria for individual risks and for the overall residual risk.
If the overall residual risk is unacceptable, the manufacturer may consider
- introduce further risk control measures to further reduce the residual risk or
- to modify the medical device or the intended purpose of the medical device.
If this is not possible, the overall residual risk remains unacceptable.
If the overall residual risk is assessed as acceptable, the manufacturer must inform users of significant residual risks and include the necessary information in the accompanying documentation to disclose these residual risks.
Chapter 9: Risk management review
The title of the chapter has changed: In the old edition it was Risk management report. But the content remains the same.
Chapter 10: Production and post-production activities
This chapter has been extensively revised. This is evident from the chapter's title: The focus is not on the information for the subsequent phases, but rather on the actions to be implemented based on the information collected and evaluated.
Furthermore, this section has been adapted with regard to Chapter 8 of ISO 13485, which deals with handling complaints, taking customer feedback into account, internal audits, control of defective products, data analysis and improvements.
10.1 General
This section emphasizes that the manufacturer must establish a system that active collects and evaluates relevant information from production and post-production phases in order to derive the right activities from it.
10.2 Information collection
This section describes in detail which information sources should be considered.
10.3 Information review
This explains how to check the information for its security relevance.
10.4 Actions
The new section “Actions” describes the measures to be applied if it is determined that information concerns safety-relevant aspects of the medical device.
On the one hand, aspects of the respective medical device are addressed, e.g.:
- Review of the risk management file
- Review of the acceptance of the residual risk
- Measures relating to products on the market
On the other hand, the entire risk management process is addressed. For example,
- to check the suitability of the RM process and
- on what changes need to be made to the process.
Annex A Rational for Requirements
This annex explains the requirements of ISO 14971 described in the normative part.
Annex B Risk management process for medical devices
Annex B contains a table of correspondence between the standards from 2007 to 2019, as well as an overview diagram of the
Risk management process for medical devices.
Annex C Fundamental risk concepts
Appendix C deals with the concepts Hazard, Sequence of events, Hazardous situations, Harm and Risk and provides examples of the terms mentioned. In the previous edition, this material was found in Appendix E. The original Appendix C is being moved to ISO TR 24971.
Conclusion:
The new ISO 14971 is more clearly worded and provides better specifications for the individual requirements. The structure of the standard and the division of the subchapters have also been improved. However, the previously very slim TR 24971 (12 pages) must now be purchased separately to receive the full scope of the old ISO 14971.
Only a few changes have been made to the content. These include, in particular:
- Special emphasis on risk-benefit analysis
- Supplementing the plan with the method and criteria for the overall risk assessment (Chapter 4.4)
- Carrying out the assessment of the overall residual risk (Chapter 8)
- Requirements for the active collection and evaluation of information from the downstream phases and emphasis on the measures to be implemented (Chapter 10)
In my opinion, ISO's goal of ensuring that established risk management processes require minimal changes to comply with the requirements of the new edition of ISO 14971 has been achieved. These clarifications will help manufacturers more easily implement the requirements in compliance with the standard.
- https://www.meddeviceonline.com/doc/a-look-at-the-iso-and-iso-tr-updates-0001 ↩
- The ISO Technical Management Board is responsible for these requirements. ↩
- ISO/IEC Guide 63:2019 Guide to the development and inclusion of aspects of safety in International Standards for medical devices ↩
- from EN ISO 14971:2019, Foreword ↩