
Medical devices have special usability requirements. Improper use of medical devices can result in harm to patients or operators. To prevent this, IEC 62366-1 defines clear processes for the usability of medical devices. The standard applies to both the EU and the US. This article focuses on Annex C of the standard, which specifies how to handle products for which there is insufficient documentation of usability according to the standard.
What is a UOUP?
The term UOUP is reminiscent of the term SOUP ("software of unknown provenance"), which is familiar from software development according to IEC 62304. Accordingly, UOUP refers to a "user interface of unknown provenance." This refers to a user interface of a medical device for which no suitable documentation exists according to IEC 62366-1.
Annex C of the standard provides manufacturers with an alternative approach, eliminating the need to implement all the activities required for usability. IEC 62366-1 defines usability engineering as part of the product development process and is not intended for re-documenting products! A user interface is considered a UOUP if:
- the product was developed before 2015 and thus before the publication of the standard (IEC 62366-1:2015).
- the product was not developed as a medical device (e.g. a computer mouse or monitor).
If the manufacturer further develops a product, all changes must be treated according to Chapters 5.1 to 5.9 of the standard. However, the unmodified parts may continue to be treated as UOUP.
What is required in the case of UOUP?
As already mentioned, IEC 62366-1 allows for an alternative path for UOUP. Similar to the approach for legacy software in IEC 62304, Annex C of IEC 62366-1 defines the process and deliverables required for UOUP. Where possible, this should build on existing documentation created during the development of the legacy user interface. This provides the manufacturer with the opportunity to avoid unnecessary re-documentation and focus on what really matters: identifying the hazards resulting from poor usability and defining corrective actions. The diagram shows the alternative path for UOUP.
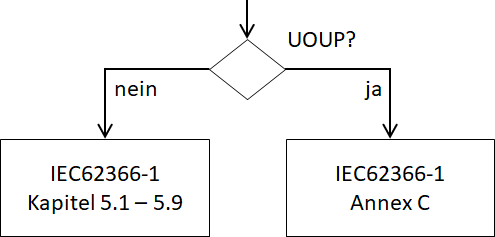
If it is a UOUP as described above, the following activities are necessary according to the table below.
| Task according to Annex C | Description |
|---|---|
| C.2.1 Use Specification | The Usability Engineering File must include the Use Specification. This must cover the following points:
|
| C.2.2 Evaluation of post-market information | The manufacturer of medical devices with UOUP should evaluate post-market information (available feedback, complaints, incidents, and reports) to identify any previously unidentified hazards. All known cases attributable to usability should be listed in the usability engineering file. The focus should be on assessing the potential severity of the harm, not the number of feedbacks, as usability errors are often not reported to the manufacturer.
In particular, the following questions should be considered (see IEC TR 62366-2:2016):
|
| C.2.3 Review hazards and hazardous situations related to usability | The manufacturer must review the risk analysis of the medical device using UOUP and ensure that the hazards and hazardous situations related to usability have been identified and documented. |
| C.2.4 Review or expand risk control measures | The manufacturer shall verify and document that appropriate risk control measures have been implemented for all hazards and hazardous situations identified in C.2.3 and that all risks are reduced to an acceptable level as indicated in the risk assessment. If necessary, risk control measures must be implemented, documented, and verified. If changes to the user interface become necessary, the changes are no longer considered UOUP, and the requirements of Chapters 5.1 – 5.9 of the standard must be explicitly applied to the changes. |
| C.2.5 Assessment of remaining risks (Residual Risk Evaluation) | The manufacturer must reassess the overall risk according to ISO 14971 and document the result either in the usability engineering file or in the risk management file. |
I highly recommend the path to usability described here to all manufacturers of medical devices with a UOUP. This will ensure that your medical device can be used safely and that the risks are controlled. Then you can continue to enjoy your UOUP in the future.
If you have any questions on this topic, I would be happy if you contact us.
Best regards
Goran Madzar