
Anyone involved in medical technology should also be familiar with the laws and regulations required for this. Therefore, today I would like to discuss the legal situation in Europe. If the thought of the MDD, the MPG, and basic requirements makes you uncomfortable, then you should read this article. You'll also learn what condoms and defibrillators have in common.
What is a medical device?
Medical devices are defined in Directive 93/42/EEC as:
Any instrument, apparatus, device, software, material or other article, used alone or in combination, including software specifically intended by the manufacturer for use for diagnostic and/or therapeutic purposes and used for the proper functioning of the medical device, which is intended by the manufacturer to be used for human beings for the following purposes:
- Detection, prevention, monitoring, treatment or alleviation of disease;
- Detection, monitoring, treatment, mitigation or compensation for injuries or disabilities;
- Investigation, replacement or modification of the anatomical structure or a physiological process;
- Birth control
and whose main intended effect in or on the human body is not affected by pharmacological or immunological Means still metabolic achieved, but whose effectiveness can be supported by such means.
If the intended use of your product is for these purposes, it is a medical device. Software (medical apps or websites) can also be considered a medical device.
MDD and MPG
There are three directives in Europe that define the legal framework for medical devices:
- Medical Device Directive (MDD) 93/42/EEC
- Directive 98/79/EC on in vitro diagnostic medical devices (IVDD)
- Directive 90/385/EEC on active implantable medical devices (AIMDD)
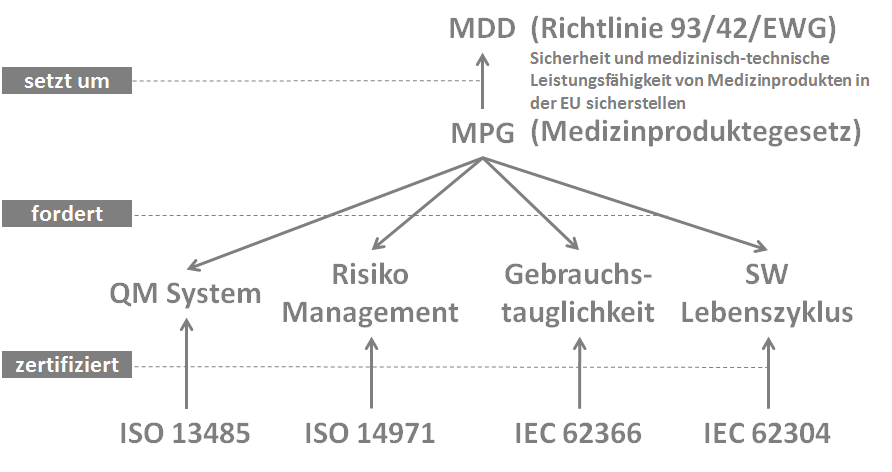
These three directives are intended to ensure the safety and medical-technical performance of medical devices in the EU. EU member states are obligated to take all necessary measures to ensure that devices may only be placed on the market and/or put into service if they meet the requirements of these directives when properly supplied, installed, maintained, and used in accordance with their intended purpose.
The Medical Devices Directive is implemented into national law through national laws. In Germany, this is done through the Medical Devices Act (MPG).
|
|
| Dipl.-Ing. Goran Madzar, Partner, Senior Systems Engineer E-mail: madzar@medtech-ingenieur.de Phone: +49 9131 691 240 |
|
Do you need support with the development of your medical device? We're happy to help! MEDtech Ingenieur GmbH offers hardware development, software development, systems engineering, mechanical development, and consulting services from a single source. Contact us. |
|
For medical device manufacturers, the MDD imposes four key requirements in the following areas:
- Quality management
- Risk management
- Software lifecycle process
- Usability
There are harmonized European standards for these four subject areas:
- EN ISO 13485 Medical devices – Quality management systems – Requirements for regulatory purposes
- EN ISO 14971 Application of risk management to medical devices
- EN 62304 Medical device software – Software lifecycle processes
- EN 62366 Application of usability to medical devices
Harmonized means that compliance with this harmonized standard also implies compliance with the essential requirements of the applicable directive. This assumption is referred to as the "presumption of conformity." Harmonized standards are developed in cooperation between the European Commission and European standardization bodies (CEN or CENELEC).
Classification of medical devices
A distinction is made active and inactive Medical devices. Active medical devices use an external energy source (electricity, accumulator, battery, thermal or kinetic energy, or gas pressure). Non-active medical devices are "passive" or powered by muscle power or gravity.
Depending on the risk of use, medical devices are divided into four classes: I, IIa, IIb and III.
The classes are defined EU-wide by Annex IX of Directive 93/42/EEC:
- Class I
- No methodological risks
- low degree of invasiveness
- no or uncritical skin contact
- temporary application ≤ 60 minutes
- Class IIa
- Application risk
- moderate degree of invasiveness
- short-term applications in the body (in the eye, intestine, in surgically created body openings)
- short-term ≤ 30 days, continuous or repeated use of the same product
- Class IIb
- Increased methodological risk
- systemic effects
- Long-term applications
- non-invasive contraception
- long-term ≥ 30 days, otherwise as for short-term
- Class III corresponds to high hazard potential
- Particularly high methodological risk
- for long-term drug delivery
- Ingredient of animal origin and in the body
- direct application to the heart, central circulatory system or central nervous system
- and of course invasive contraception
Examples of medical devices belonging to the different classes are shown in the table below.
| Class I | Class IIa | Class IIb | Class III |
| medical instruments | Dental materials | Anesthesia machines | cardiac catheter |
| assistants | Disinfectants (for instruments and equipment) | Ventilators | artificial hip, knee or shoulder joints |
| wheelchairs | diagnostic ultrasound devices | Irradiation devices | Stents |
| Nursing beds | Disposable syringes | Blood bag | absorbable surgical suture material |
| Support stockings | Hearing aids | Defibrillators | Intrauterine device (IUD) |
| dressings | Contact lenses | Dialysis machines | breast implant |
| reusable surgical instruments | PACS | Condoms | pacemaker |
| Surgical textiles | Tracheal tubes | Contact lens cleaner |
Classification is based on intended use. Did you know that condoms and defibrillators are classified the same? I find that a bit amusing.
Conformity assessment
The conformity assessment is carried out by the manufacturer. The risk class plays an important role in the conformity assessment. For a Class I product, a declaration of conformity according to Annex VII is sufficient. To do this, the manufacturer is obliged to provide evidence of compliance with the essential requirements by means of technical documentation. The manufacturer must also be able to monitor the product after it has been placed on the market and initiate corrective measures. The involvement of the notified body is not necessary in this case. The situation is different for the higher risk classes (IIa, IIb and III). In these cases, a conformity assessment procedure according to Annex II can be carried out. For this, the manufacturer must establish a complete quality assurance system (usually according to ISO 13485). This is regularly certified and audited by notified bodies in audits. Another option is to have the development carried out via an EC type-examination according to Annex III. The notified body checks compliance with the essential requirements using a type and the associated documents. It must then be ensured that the manufacturer is able to manufacture the product correctly. This can be achieved through an EC verification according to Annex IV, in which the finished products are submitted to the notified body for testing. It is also possible to have the quality assurance of production certified according to Annex V or the quality assurance of the product certified according to Annex VI by a notified body.
CE marking
After a successful conformity assessment procedure, a CE marking may be affixed to the product and thus placed on the European market. If a notified body is involved in the conformity assessment procedure, its four-digit identification number is indicated after the marking.
Notified bodies (engl. Notified BodiesNotified Bodies (NBBs) are government-authorized bodies that – depending on the risk class of the medical devices – carry out tests and assessments as part of the conformity assessment to be carried out by the manufacturer and certify their accuracy according to uniform assessment standards. Manufacturers can contact a notified body of their choice that is designated for the relevant procedure and product category.
Conclusion
I hope the laws and guidelines have become a little clearer to you. This article doesn't claim to cover all topics completely and comprehensively. The goal was to explain the basic concepts. Therefore, for this article, the following applies: "For risks and side effects, read the guidelines and laws or contact your quality manager :-)"
I welcome feedback and would love for you to contact me. Feel free to leave a comment on the article. If you know someone who might also be interested in the blog, I'd be very happy if you would recommend it.
Best regards
Goran Madzar